demoHAP1
Demo for hap1 cells is provided with the package. There are two bash scripts provided inside the demo/demo2_hap1_cell/. First one is run_hap1_preprocess.sh and second is run_hap1_cell where run_hap1_preprocess.sh should be run first. Both are explained as under.
Step 1: dds_analysis preprocess
The dds_analysis preprocess step prepares the input files for further analysis using the dds_analysis module. It requires specifying various parameters and input file paths. Here is the code:
# Before running following steps, it assumes that DMRs are already predicted by dmr_analysis
dds_analysis preprocess \
-in_folder ../../data/hap1_cell/in_data/final_demo_data/hap1_cell/out_data/DMR_CpG_context/out_map2genome/ \
-in_string '_hap1' \
-in_tss_file_mr ../../data/hap1_cell/in_data/final_demo_data/hap1_cell/out_data/DMR_CpG_context/out_map2genome/3_chroms_all_mr_data_range_dmrRanking_TSS_Up5000_Down1000_removedShort_overlap1e-09.bed \
-in_dist_file ../../data/hap1_cell/in_data/final_demo_data/hap1_cell/out_data/DMR_CpG_context/out_map2genome/3_chroms_all_mr_data_range_dmrRanking_noGenes_5dist_Up1000000_Up5000removedShort_overlap1e-09.bed \
-in_deg_file ../../data/hap1_cell/in_data/final_demo_data/hap1_cell/in_data/DEG/HAP1_P1_vs_HAP1_KO1_differentially_expressed_genes_min1.1Fd_min1RPKM.txt \
-out_folder ../../data/hap1_cell/out_data/ \
-tss_file ../../data/hap1_cell/in_data/final_demo_data/hap1_cell/out_data/DMR_CpG_context/data/TSS_Up5000_Down1000_removedShort.bed \
-full_mr_file ../../data/hap1_cell/in_data/final_demo_data/hap1_cell/out_data/DMR_CpG_context/3_chroms_all_mr_data_range_dmrRanking.bed \
-in_genome_file ../../data/hap1_cell/in_data//final_demo_data/genome/hg38/hg38_all_enhancers_merged_hglft_genome_327b3_4dmr.bed \
-gene_col_name '#gene'
echo "To find DMR regions that are overlapping with TSS or 5distance regions of DEG - and preprocess Done"
Here head of MR file with TSS region used as input is displayed: 3_chroms_all_mr_data_range_dmrRanking_TSS_Up5000_Down1000_removedShort_overlap1e-09.bed
chr3 8500658 8506658 chr3:8500658:8506658:NR_033378||TSS:5000:1000||LMCD1-AS1:-:8221146:8501658 chr3 8462210 8501263 chr3:mr61:hypo:U 0.009313857563925538
chr3 8500658 8506658 chr3:8500658:8506658:NR_033378||TSS:5000:1000||LMCD1-AS1:-:8221146:8501658 chr3 8502690 8519521 chr3:mr62:hypo:D 0.9473746593032959
chr3 8496806 8502806 chr3:8496806:8502806:NM_001278234||TSS:5000:1000||LMCD1:+:8501806:8568125 chr3 8462210 8501263 chr3:mr61:hypo:U 0.009313857563925538
chr3 8496806 8502806 chr3:8496806:8502806:NM_001278234||TSS:5000:1000||LMCD1:+:8501806:8568125 chr3 8502690 8519521 chr3:mr62:hypo:D 0.9473746593032959
chr3 8496822 8502822 chr3:8496822:8502822:NM_001278233&NM_001278235||TSS:5000:1000||LMCD1:+:8501822:8574668&8551274 chr3 8462210 8501263 chr3:mr61:hypo:U 0.009313857563925538
chr3 8496822 8502822 chr3:8496822:8502822:NM_001278233&NM_001278235||TSS:5000:1000||LMCD1:+:8501822:8574668&8551274 chr3 8502690 8519521 chr3:mr62:hypo:D 0.9473746593032959
While head of methylation regions file overlapped with 5 dist region is following: 3_chroms_all_mr_data_range_dmrRanking_noGenes_5dist_Up1000000_Up5000removedShort_overlap1e-09.bed
chr3 7540284 8535284 chr3:7540284:8535284:NR_046606||5dist:5000:1000000||GRM7-AS1:-:7519740:7535284 chr3 8101574 8101679 chr3:mr0:mix:U 0.0020392338824331358
chr3 7540284 8535284 chr3:7540284:8535284:NR_046606||5dist:5000:1000000||GRM7-AS1:-:7519740:7535284 chr3 8102773 8107735 chr3:mr1:mix:D 0.9863575510990631
chr3 7540284 8535284 chr3:7540284:8535284:NR_046606||5dist:5000:1000000||GRM7-AS1:-:7519740:7535284 chr3 8119920 8125713 chr3:mr3:mix:D 0.9800274651202444
chr3 7540284 8535284 chr3:7540284:8535284:NR_046606||5dist:5000:1000000||GRM7-AS1:-:7519740:7535284 chr3 8136677 8139855 chr3:mr4:mix:D 0.9795108977746336
Input file that consist of methylation regions is following: 3_chroms_all_mr_data_range_dmrRanking.bed
chr3 8101574 8101679 chr3:mr0:mix:U 0.0020392338824331358
chr3 8102773 8107735 chr3:mr1:mix:D 0.9863575510990631
chr3 8108750 8115603 chr3:mr2:mix:D 0.9763066966833287
chr3 8119920 8125713 chr3:mr3:mix:D 0.9800274651202444
chr3 8136677 8139855 chr3:mr4:mix:D 0.9795108977746336
Differentially expression file is below: HAP1_P1_vs_HAP1_KO1_differentially_expressed_genes_min1.1Fd_min1RPKM.txt
AADAT 0.006070520526336222 16.90556267502578 15.3097562941675 16.90556267502578 13.389386678473848 12.573766472283713 11.850516581141726
AARS 0.0006937443220722942 58.2440643381434 58.810394758261374 59.96440144545457 33.27901746502996 32.504309785698375 29.03704380096956
AATK 0.02271698518484375 2.5554957565029683 2.643258236240186 2.0719454919445686 3.1558313097737094 3.4185648937579978 3.93619336697585
ABAT 1.9842909957276316e-05 0.36312747169342474 0.4141701884134888 0.4404845359100948 1.1351009276855188 1.1024911071781391 1.1728150315920693
ABCA1 0.013656558666489764 3.074263544209392 2.5879886642784986 2.4299433764226444 1.6758838422325957 1.4414555073263386 1.3930064239495081
Set paths:
#main path of input data
IN_DATA_PATH='../../data/hap1_cell/in_data/final_demo_data/hap1_cell/'
#path of DMR results from dmr_analysis
IN_MR_PATH=${IN_DATA_PATH}'/out_data/DMR_CpG_context/'
#path of DEG results from bpb3
IN_DEG_PATH=${IN_DATA_PATH}'/in_data/DEG/'
#DEG file name from bpb3 differential_analysis, the original DEF file from bpb3 that was used to convert Zscores in dds_analysis preprocess
IN_DEG_FILE='HAP1_P1_vs_HAP1_KO1_differentially_expressed_genes_min0Fd_min0RPKM.txt '
in_data_str='_hap1'
#path to output data
OUT_PATH='../../data/hap1_cell/out_data/'
#path to exported MRs that are not located in TSS or enhancer regions
FILE_FOLD=${OUT_PATH}/out4mr_not_in_tss_enhancer
#file name for background sample list that contain all MRs not located in TSS or enhancers
BACK_FILE=${OUT_PATH}/background_samples_list.tsv
#whether to skip below two steps in the pipeline
is_run_dmr_export=1 # 1 for exporting, other values for skipping this step
is_run_dtarget=1 # 1 for run dTarget prediction , other values for skipping this step
Step 2: DMR data export
The DMR data export step involves exporting DMR data that is either located in TSS or 5’distance regions. It uses the dmr_exportData command from the dmr_analysis module. Here is the code:
# Before running this step, ensure DMRs are already predicted by dmr_analysis
dmr_analysis dmr_exportData \
--input_mr_data_folder ${IN_MR_PATH} \
--output_file_folder ${OUT_PATH}/out4dmr_in_deg_tss_5dist \
--input_file_format 0 \
--number_of_processes 10 --input_file ${OUT_PATH}/uqdmr_regions_in_deg_tss_5dist${in_data_str}.bed -wtStr 'HAP1_P_'
echo "Export data of DMRs overlapping to TSS or 5distance - Done"
chr13 95217448 95424140 chr13:mr8:hypo:D chr13:95300446:95306446:NM_005845||TSS:5000:1000||ABCC4:-:95019828:95301446;chr13:95300451:95306451:NM_001301829&NM_001105515||TSS:5000:1000||ABCC4:-:95019834&95095770:95301451
chr3 9055959 9261176 chr3:mr88:mix:D chr3:9248646:9254646:NM_014850||TSS:5000:1000||SRGAP3:-:8980590:9249646
# Export data of MRs not in TSS or enhancers
dmr_analysis dmr_exportData \
--input_mr_data_folder ${IN_MR_PATH} \
--output_file_folder ${OUT_PATH}/out4background \
--input_file_format 0 \
--number_of_processes 10 --input_file ${OUT_PATH}/uqdmr_regions_not_in_deg_tss_5dist${in_data_str}.bed -wtStr 'HAP1_P_'
echo "Export data of MRs not in TSS or enhancers - Done"
Step 3: Background file list creation
The background file list creation step involves creating a background file list if it doesn’t already exist. The background file list contains all MRs that are not located in TSS or enhancer regions. Here is the code:
background_filelist="${OUT_PATH}/backgroundFileList.txt"
if [[ ! -f "$background_filelist" ]]; then
cd ${OUT_PATH}/out4background
ls | grep ".bed" > $background_filelist
cd -
echo "Creating a list of background files - Done"
else
echo "Background file list already exists."
fi
echo "Background file list creation - Done"
Step 4: DEG File preparation:
Prepare a tab delimited gene expression file in which the group mean values and rratio are added. This file will be used to plot average methylation levels of selected gene in TSS and Enhancer regions After inputting a DEG file exported by bpb3 differential_expression, it exports a tab delimited file by adding three columns values of the group mean and rratio. This filtered DEG file will only be used in plot_tss_enhancer_mrs for exporting data This function only consider input data as RPKM values
gene_mr_file=${OUT_PATH}/uqGeneDmr_regions_in_deg_tss${in_data_str}.bed
dds_analysis filterDEG4bpb3 --in_group1_str 'HAP1_P1' --in_group2_str 'HAP1_KO1' \
--in_folder ${IN_DEG_PATH} \
--in_file ${IN_DEG_FILE} \
--min_median_RPKM 0 --rr_cutoff 0.0
#we can skip this manual input step if know the input gene expression file name
if [ 1 == 2 ];
then
read -p "To continue please copy the exported zscore cluster file name and path from bpb3 filterDEG4bpb3 then click return: " gene_exp_file
echo "gene_exp_file is : $gene_exp_file "
read -p "To continue please copy the exported group mean file name and path from bpb3 filterDEG4bpb3 then click return: " IN_DEG_FILE
echo "IN_DEG_FILE is : $IN_DEG_FILE "
fi
#end test
We assume know the input gene exp file name gene_exp_file0=${IN_DEG_PATH}/${IN_DEG_FILE} Here we assume file name end with .txt
finds='.txt'
replace1='_rratio_filtered4cluster.csv'
replace2='_rratio_filtered.csv'
gene_exp_file=${gene_exp_file0//$finds/$replace1}
IN_DEG_FILE=${gene_exp_file0//$finds/$replace2}
echo ""
echo "gene_exp_file is : $gene_exp_file "
echo ""
echo "IN_DEG_FILE is : $IN_DEG_FILE "
echo ""
# path of DMRs associated with DEG, TSS and 5distance, prepared by run dds_analysis preprocess
in_mr_data_folder=${OUT_PATH}/out4dmr_in_deg_tss_5dist
# a file for a list of background samples
in_background_mr_file=$BACK_FILE
#number of random sampling for the test
number_of_samples=10
HAP1_P1_vs_HAP1_KO1_differentially_expressed_genes_min1.1Fd_min1RPKM_rratio_filtered4cluster.txt head is following:
FAM166AP3 -0.4982081983511595 -0.9394358401186298 -0.6707136220439952 -1.7706649617423529 -1.7471009146010719 -1.759213053860002
RP11-561I11.3 -0.7538360859791011 -0.8163195171038196 -1.0486517498078607 -1.7688207617054021 -1.745213065641895 -1.7573721462587126
AC009299.4 -1.240842456343193 -0.9978063637216715 -0.8863905055414616 -1.7671318296845058 -1.743530972980885 -1.7556519670664428
FAM41C -1.1073411320948858 -1.332098180960522 -1.106948209449783 -1.7284061127409744 -1.7596852452138803 -1.7717564601163207
ARSE -0.8784524519818689 -1.0690546795243978 -0.8785276236434105 -1.7487097864101793 -1.7626589252195035 -1.639011789153589
RP11-442H21.2 0.05741831209072534 -0.13825346076745668 -0.22036980044826926 -1.3796771543980877 -1.529168641624075 -1.7594854289267512
RP11-422P24.12 -1.096537825707665 -1.0424048390869258 -1.282182441903589 -1.7668478469733657 -1.7432581180749236 -1.6746361853756075
RP4-590F24.2 -1.1047323074565782 -1.228979590661412 -1.1307512157539712 -1.676088854525317 -1.760365854510401 -1.755233404634837
MIR4419A -0.16271440608073698 -0.16705481613532722 -0.49940830855525836 -1.582622407034913 -1.545955456168695 -1.5599446568981838
INHBE 0.4194153086557727 0.37903743335847656 0.4158868327542566 -1.3577006540490815 -1.153783055168014 -1.4861394387459987
CHAC1 2.3416693314259813 2.3286405094448774 2.3462661518248256 0.2075086031948541 0.21713102593121023 0.05283409591964295
Step 5: dTarget Prediction based in expression profiles:
The dTarget prediction step predicts putative target genes for DMRs based on gene expression profiles. It performs the prediction separately for DMRs associated with TSS regions and 5’distance regions. Here is the code:
# Predict target genes for DMRs overlapping with TSS regions
dds_analysis dTarget_methy_vs_express -inGeneMRfile $gene_mr_file -mrTAB \
-inGeneEXPfile $gene_exp_file -expTAB \
-inMRfolder $in_mr_data_folder -outName 'tss_region_' \
-output_path $OUT_PATH -sampleName sample_name4replace.tsv \
-pathDepth 1 -inBackgroundList $in_background_mr_file -reg_cutoff 0.5 -cutoff 1.0 -totalSamples $number_of_samples -numOfprocesses 10
echo "Done with TSS target gene prediction"
echo "Target gene prediction for DMRs overlapping with TSS regions - Done"
# Predict target genes for DMRs associated with 5'distance regions
dds_analysis dTarget_methy_vs_express -inGeneMRfile $gene_mr_file -mrTAB \
-inGeneEXPfile $gene_exp_file -expTAB \
-inMRfolder $in_mr_data_folder -outName 'distance_region_' \
-output_path $OUT_PATH -sampleName sample_name4replace.tsv \
-pathDepth 1 -inBackgroundList $in_background_mr_file -reg_cutoff 0.5 -cutoff 1.0 -totalSamples $number_of_samples -numOfprocesses 10
echo “Done with 5distance region target gene prediction”
echo “Target gene prediction for DMRs associated with 5’distance regions - Done”
Step 6: Plotting selected target gene and DMR associations
This step involves plotting the associations between selected target genes and DMRs based on gene expression profiles. Here is the code:
echo ${gene_exp_file}
echo ${OUT_PATH}/out4dmr_in_deg_tss_5dist
dds_analysis plot_mr_vs_exp -inGeneEXPfile ${gene_exp_file} \
-dpi 300 -inMRfolder ${OUT_PATH}/out4dmr_in_deg_tss_5dist \
-expTAB -inGene 'TRIM32' -inMR 'chr9:mr104' -wtStr 'HAP1_P' -output_path ${OUT_PATH}
echo "Done with plot_mr_vs_exp "
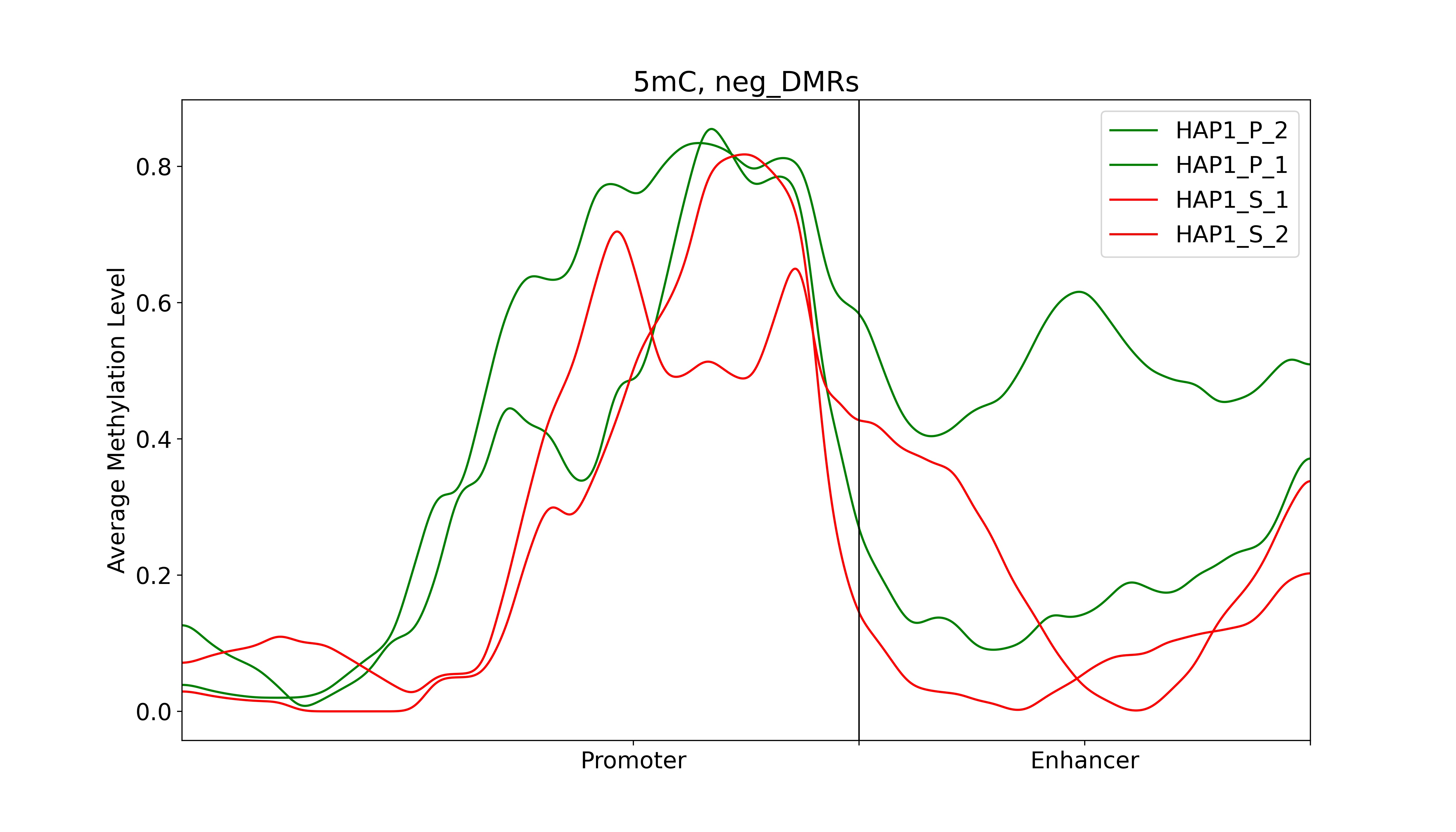
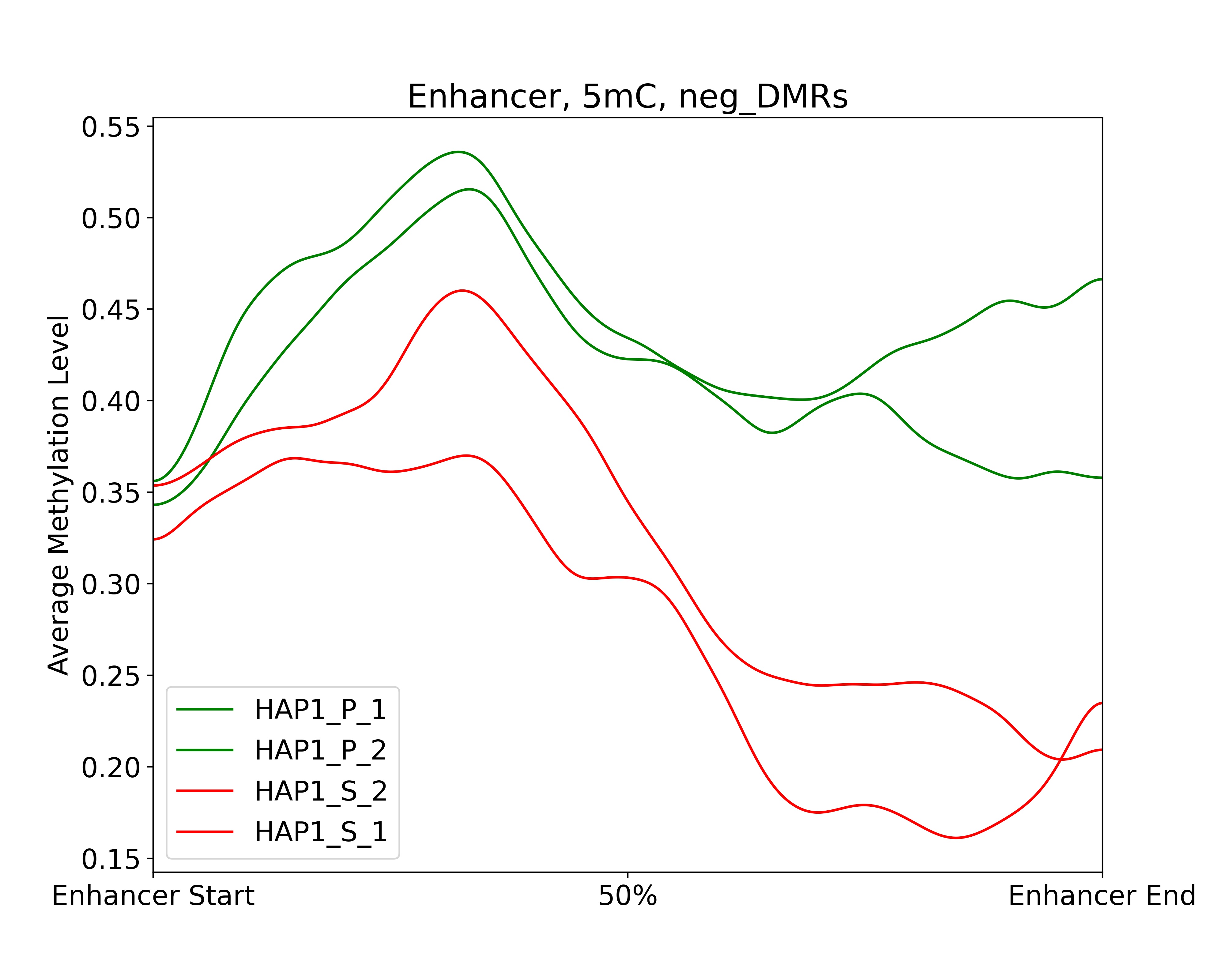
Step 7: Plotting average methylation pattern
The final step involves plotting the average methylation in TSS and enhancer regions for selected target gene. Here is the code:
dds_analysis plot_tss_enhancer_mrs \
-exp_file $IN_DEG_FILE \
-dmr_file ${IN_MR_PATH}/3_chroms_all_mr_data_range_dmrRanking.bed \
-tss_file ${OUT_PATH}/tss_region_10sampling.csv \
-enc_file ${OUT_PATH}/distance_region_10sampling.csv \
-is_negative 1 -genes 'MRPS25,PLCL2,TRIM32' -mr_folder ${OUT_PATH}/out4dmr_in_deg_tss_5dist/ \
-folder_name '' --dmr_file_not_compressed \
-gX 2000 -gY 1000 -wtStr 'HAP1_P_' \
-out_folder ${OUT_PATH}/plot_tss_enhancer_mrs
echo "Done with plot_tss_enhancer_mrs"